
In computational chemistry, evaluating the results of molecular docking is essential for drug development and understanding biochemical interactions. The PoseBusters suite provides a comprehensive toolkit for assessing docking poses generated by both physics-based and AI/ML-based methods. Here, we outline how PoseBusters is helping us evaluating the results of physics-based and AI/ML-based docking methods, where Root Mean Square Deviation (RMSD) alone is not sufficient.
RMSD: A Common but Insufficient Metric
Root Mean Square Deviation (RMSD) is a commonly used metric in molecular docking to measure the similarity between the predicted pose and a reference pose. While RMSD is useful for quantifying the overall geometric deviation, it has limitations:
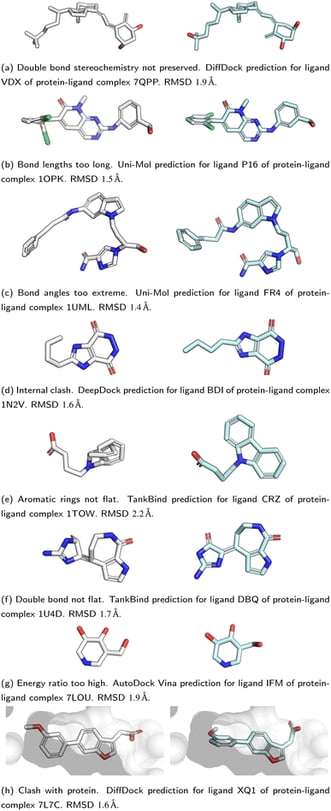
- Lack of Chemical Insight: RMSD does not account for chemical validity, such as proper bond orders, valency, and stereochemistry.
- Global Measure: RMSD provides a global measure of similarity but can miss critical local inaccuracies in bond lengths, angles, and other structural features.
- Energetic Considerations: RMSD does not consider the energetic feasibility of the predicted pose. The molecule might be contorted into a conformation with high energy, leading to instability or unrealistic reactivity.
1. Chemical Validity and Consistency
a. Input Loading
The molecular structure is loaded into RDKit, a robust cheminformatics software, ensuring it can be processed without errors. This step establishes a reliable foundation for subsequent validation checks.
b. Sanitization Checks
RDKit performs a series of sanitization checks to confirm chemical validity. These checks verify:
- Bond Orders: Ensuring the number of bonds between atoms is chemically feasible.
- Atom Valency: Confirming each atom forms the correct number of bonds according to its chemical properties.
These checks prevent chemically impossible configurations from advancing further.
2. Molecular Formula and Bonds
The molecular formula and bond types of the input molecule are cross-referenced against the known true structure. This step ensures that the molecular formula (atom count and type) and bond types (single, double, etc.) are accurate and consistent with expected chemical properties.
3. Stereochemistry Checks
Stereochemistry refers to the 3D spatial arrangement of atoms in a molecule. PoseBusters performs specific checks for:
- Tetrahedral Chirality: Verifying the spatial arrangement around a central atom with four substituents.
- Double Bond Stereochemistry: Checking the geometric configuration around double bonds.
These checks ensure that the molecule’s 3D structure matches the expected configuration.
4. Bond Lengths and Angles
PoseBusters measures bond lengths and angles within the molecule, ensuring they fall within specific tolerance limits that reflect realistic chemical structures. These measurements are assessed using distance geometry bounds from RDKit, confirming that the molecule’s geometry is physically plausible.
5. Planarity of Aromatic Rings and Double Bonds
The suite checks for the planarity of atoms in aromatic rings and double bonds. These structures are expected to lie within a plane, supporting their stability and chemical reactivity. Ensuring planarity is crucial for the proper functioning of aromatic systems and conjugated double bonds.
6. Internal Steric Clash
PoseBusters calculates the distance between all pairs of non-bonded atoms in the molecule. It ensures these distances are above a minimum threshold, preventing internal steric clashes that could destabilize the molecule.
7. Energy Ratio
The energy of the predicted conformation is compared to a reference set of conformations generated by RDKit. The pose must not exceed an energy ratio threshold, ensuring it is energetically reasonable compared to typical conformations. This check prevents the consideration of energetically unfavorable poses.
8. Intermolecular Validity
a. Minimum Distances
PoseBusters checks the minimum distances between the ligand and protein or cofactors to ensure no physically implausible overlaps or contacts occur.
b. Volume Overlap
The overlapping volume between the ligand and surrounding molecules (such as proteins and cofactors) is calculated. This overlap must be below a set threshold to avoid unrealistic packing, ensuring the ligand fits well within the binding site.
Conclusion
PoseBusters applies rigorous criteria to evaluate each ligand pose generated by docking algorithms, addressing the limitations of RMSD by incorporating chemical validity, stereochemistry, geometric accuracy, and energetic feasibility. Only poses that pass all these tests are considered "PB-valid," indicating they are both physically plausible and chemically accurate. This comprehensive assessment ensures that only the most reliable poses are considered successful, helping to identify and improve the predictive capabilities of docking algorithms. By employing the PoseBusters suite, researchers can confidently evaluate and refine their docking results, paving the way for more accurate and effective drug discovery efforts.
Resources
Paper: PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences
GitHub: PoseBusters: Plausibility checks for generated molecule poses
PyPI: https://pypi.org/project/posebusters/
Documentation: https://posebusters.readthedocs.io/en/latest/
.svg)
.svg)
.svg)